|
Greener Journal of Medical Sciences
Vol. 12(1), pp. 156-160, 2022
ISSN: 2276-7797
Copyright ©2022, the copyright of this article is
retained by the author(s)
https://gjournals.org/GJMS
|

|
Acrania:
Report of a Rare Congenital Malformation
Okagua KE1;
Okagua J2; Eli S3; Eke CM4
Department of Obstetrics & Gynaecology,
Rivers State University Teaching Hospital1
Department of Paediatrics, University of Port
Harcourt Teaching Hospital2
Mother, Baby and Adolescent Care Global
Foundation3
Image Diagnostic Centre4
|
ARTICLE INFO
|
ABSTRACT
|
|
Article No.: 050422044
Type: Research
Full Text: PDF, HTML, PHP, EPUB
|
Acrania is a rare lethal congenital anomaly
in which there is partial or complete absence of the fetal scalp bones
(calvarium). The brain tissue is completely but abnormally developed.
We present a 22-year-old primigravida, an
unmarried secondary school drop-out who presented for prenatal care at 31
weeks of gestation and developed pre-term prelabor rupture of membranes a
week later. An obstetric scan at presentation revealed acrania of the fetus
and she was offered medical termination of the pregnancy which was declined
insisting on awaiting fetal maturity. She was managed conservatively until
34weeks of gestation when she had a lower segment caesarean section. The
Outcome was a live 1.8kg female fetus with acrania and other malformations
who suffered an Early Neonatal Death.
Her late antenatal booking deprived her of
an early diagnosis when counselling for medical termination would have been
more acceptable while her religious background gave her optimism for a
favourable outcome despite adequate counselling on the prognosis for this
very rare lethal medical condition diagnosed in utero.
|
|
Accepted: 05/05/2022
Published: 11/05/2022
|
|
*Corresponding Author
Dr Kenneth E. Okagua, (MBBS, FWACS, FICS).
E-mail: kokagua@ hotmail.com
|
|
Keywords: acrania,
rare, congenital, malformation.
|
|
|
|
INTRODUCTION
Acrania is a rare lethal congenital anomaly
in which there is partial or complete absence of the fetal scalp bones (calvarium).1,2
The brain tissue is completely but abnormally developed unlike in anencephaly.2
It is frequently confused with anencephaly. Most cases are diagnosed by 1st
trimester ultrasound scan.
It is extremely rare and its sporadic nature
suggests a low recurrence risk.2Only 6 cases had been reported in
the literature1 by 1996 with 5 of them diagnosed by sonography in
the first trimester and terminated electively while one is the first known
surviving case`. Few other caseshave been reported since then largely not surviving
beyond infancy.3,4
The first known surviving case was the child
of a 29 year old Japanese woman delivered at 38 weeks gestation by vaginal
delivery as desired by the patient despite full knowledge of the risk of fetal
death.1 The outcome was a male 2.47kg infant with Apgar scores of 4
at 1minute and 2 at 5 minutes. He had scalp and dural defect, subarachnoid
haemorrhage, cerebrospinal fluid leakage and partial cerebral contusion. He was
observed without resuscitation and 10mins after delivery his respiration and
general condition improved. At the request of his parents he subsequently
underwent repair of the scalp defect and cerebrospinal fluid leakage and at 3
months of age had a sub-duro-peritoneal shunt for hydrocephalus. He was
severely retarded with a developmental quotient of 10 at 3 years.1,3
CASE
REPORT
Ms ON is a 22 year
old primigravida, an unmarried secondary school drop-out, who was registered
for antenatal care in our facility at 31 weeks gestation. She was brought by
her church pastors’ wife who was responsible for her bills.
Her pregnancy was
undesired. She attempted termination of the pregnancy at about 6 weeks GA with
herbal drugs and quinine tablets. Her pregnancy had subsequently been
uneventful until she presented to us for antenatal care. She had received 2
doses of tetanus toxoid from a midwife who offered her some form of antenatal
care prior to presentation. Her booking parameters where a blood pressure of
90/60mmHg; weight of 56kg; blood group was O rhesus ‘D’ positive; haemoglobin
genotype was AA; packed cell volume was 36% and she was sero-negative for
hepatitis B surface antigen and HIV I&II. Her Venereal Disease Research
Laboratory (VDRL) test was non-reactive. Her urine was negative for protein and
glucose.
Her menarche was at
15 years of age. She menstruated for 4 days in a regular monthly cycle. She had
no menstrual abnormalities. She was aware of contraception but did not practice
any.She was the second child in a family of 2 children, both girls. Her mother died
of an unknown illness when she was a teenager and her father was unemployed.She
had no family history of chronic medical illness. She was an unmarried
secondary school drop-out. She was impregnated by her teenage boyfriend who
denied paternity. She denied consumption of alcoholic beverages nor tobacco
products.
A week after
registration for antenatal care, she presented to the labour ward, at 32 weeks
gestation, with complaints of gush of fluid per vaginam of 4 hours duration.
There was no associated abdominal pain nor bleeding per vaginam. There was no
history of trauma or other constitutional symptoms.
On examination, the
feto-maternal vital signs were within normal limits and liquor drainage was
confirmed on sterile speculum examination. There was no cord prolapse. A full
blood count and C-reactive protein were not suggestive of overt/occult
infection.
An obstetric
ultrasound scan done on admission revealed a single active fetus in utero, in
longitudinal lie and cephalic presentation. The biophysical profile score was
8/12, the maturity was 32 weeks of gestation and the estimated fetal weight was
1.8kg. Organ survey revealed no gross anomaly of the cardiovascular,
gastrointestinal and urogenital systems. There was however absent cranial
bones, exaggerated brain matter and asymmetry of the normal spinal lordosis.
There was moderate oligohydramnios. The placenta was posterio- fundal in
position. The cervical os was mildly dilated. There were no co-existing masses.
A diagnosis of
preterm prelabour rupture of fetal membranes with congenital malformations
including Acrania was made. The diagnosis, the fetal prognosis and the need for
medical termination of the pregnancy was explained to the patient but she
declined consent because she was hopeful of a favourable outcome despite
adequate counselling and had the active support of her pastor. She was admitted
into the maternity ward, placed on strict bed rest, received prophylactic
antibiotics and parenteral dexamethasone to aid lung maturity.
She was managed conservatively
until 34 weeks of gestation when she was offered a lower segment Caesarean
Section. Intra-operative findings were a well formed lower uterine segment;a
live female 1.8kg infant in cephalic presentation, APGAR scores were 41,05
with multiple congenital anomalies including absent fetal skull bones, prominent
brain matter, absent nose, absent forearms/fingers and prominent toe on both
lower limbs; the placenta was fundal and weighed 400g; the ovaries, tubes and
bladder appeared normal; there was minimal amniotic fluid and estimated blood
loss was 400mls. The fetus made a few gasps of breath and died within 5 minutes
of delivery.
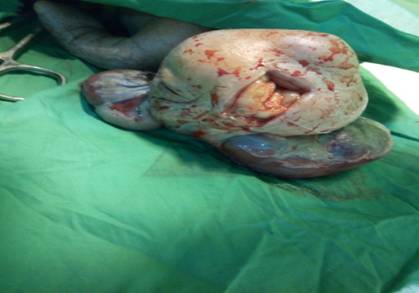
Figure 1:
Cranial and Facial anomalies
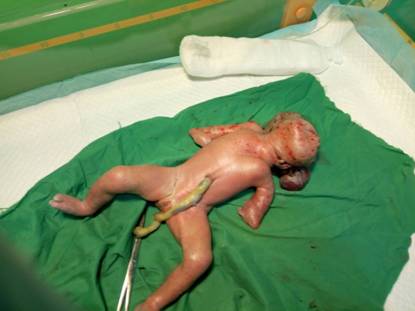
Figure 2:
Limb anomalies
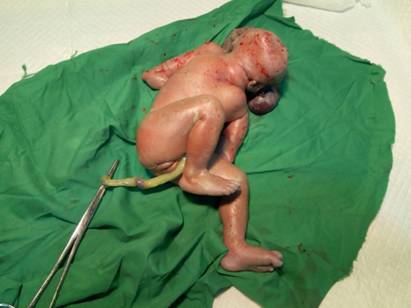
Figure 3:
Lower limb anomalies
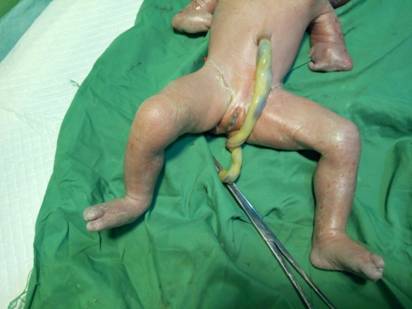
Figure 4:
Genitalia
Post operatively, she received prophylactic
antibiotics and analgesics. She also received intravenous 5% dextrose saline
for the 1st 24 hours and thereafter commenced on graded oral sips
and progressed to fluid and normal diet. Her post operative period was
uneventful, she received further counselling and was discharged home on the 5th
post operative day in satisfactory clinical condition. She was seen in the post
natal clinic after 6 weeks and was in satisfactory clinical condition. The
surgical site was healed and the uterus had involuted. She had resumed
menstruation. She was further counselled on the diagnosis and family planning
and was discharged from the clinic.
DISCUSSION
Acrania is extremely rare and its
sporadic nature suggests a low recurrence risk.2 Patient counselling
is difficult because there is no evidence of specific genetic origin.2
It is often difficult to distinguish between anencephaly, acrania and amniotic
band sequence prenatally5and postnatal differentiation as was done in
our patient is imperative. Amniotic band syndrome is a collection of fetal
malformations associated with fibrous bands that appear to entrap or entangle
various fetal parts in utero and can affect any organ or system and cause a
single or multiple anomalies.6,10
The diagnosis of acrania can be established
sonographically in the first trimester if a large mass of disorganised brain
tissue covered only by a thin membrane is detected.7,9
Our patients’ late antenatal
registration which is common with teenage pregnancies excluded early diagnosis
of her condition at a gestational age when medical termination of the pregnancy
may have been acceptable. Her situation was further compounded by her religious
background of being sponsored by her pastor who is unlikely to accept medical
termination of the pregnancy. She thus ended up with a caesarean section scar
for a fetus with minimal chance of survival.
Continuous advocacy onthe need to
minimize out of school children, female education, provision of youth friendly contraceptive
services,early antenatal registration of teenage pregnancy and re-orientation
of religious bodies on medically indicated interventions will improve outcomes
and avoid unnecessary surgical interventions.
REFERENCES
1.
Kuramata H, Tamaki N, Sawa H et al. Acrania:
Report of the First Surviving Case. Pediatr Neurosurg 1996; 24:52-54.
2.
Aman DM. A Rare Case Report of Acrania. Bang.
J Neurosurgery 2021;11(1):50-53.
3.
Bianca S, Ingegnosi C, Auditore S et al.
Prenatal and postnatal findings of acrania. Arch Gynaecol Obstet 2005; 271: 257
-259.
4.
Ouma JR. Acalvaria – report of a case and
discussion of the literature. Br J Neurosurg 2019; 33(2): 224 – 225.
5.
Hawasli AH, Beaumont TL, Vogel TW et al.
Acalvaria. J Neurosurg Pediatr 2014; 14(2): 200 – 202.
6.
Harrington BJ, Horger EO, Edwards JG. A
counselling dilemma involving anencephaly, acrania and amniotic bands. Genet Couns
1992; 3(4): 183 – 186
7.
Cincore V, Ninios AP, Pavlik J et al.
Prenatal diagnosis of acrania associated with amniotic band syndrome. Obstet
Gynaecol 2003; 102 (5 Pt 2): 1176 – 1178.
8.
Weissman A, Diukman R, Auslender R. Fetal
acrania: five new cases and review of the literature. J Clin Ultrasound 1997;
25(9): 511 – 514.
9.
Harris CP et al. Acalvaria: a uniqe
congenital anomaly. Am J Med Genet 1993;
46: 694 – 699.
10.
Yang YC, W CH, Chang FM et al. Early prenatal
diagnosis of acrania by transvaginal ultrasonography. J Clin Ultrasound 1992;
20: 343 – 345.
|
Cite this Article: Okagua KE;
Okagua J; Eli S; Eke CM (2022). Acrania: Report of a Rare Congenital
Malformation. Greener Journal of Medical
Sciences, 12(1): 156-160.
|